Introduction
The KnotProt 2.0 database (the updated version of the KnotProt database) collects information about entangled proteins, i.e. proteins which form knots, slipknots, and knotoids. Such structures are defined in either probabilistic, or deterministic and are shown in Fig. 1 (based on disulfide or ion bonds) way (the first version of the KnotProt analyzed only proteins with probabilistic knots and slipknots). The KnotProt 2.0 database classifies all such proteins, represents their entire complexity in the form of a “knotting fingerprint” [1] (motivated by work [2]), and presents many biological and geometrical statistics based on these results. The KnotProt 2.0 is a self-updating database based on protein chains deposited in Protein Data Bank (PDB) and currently contains more than 2000 entangled structures.
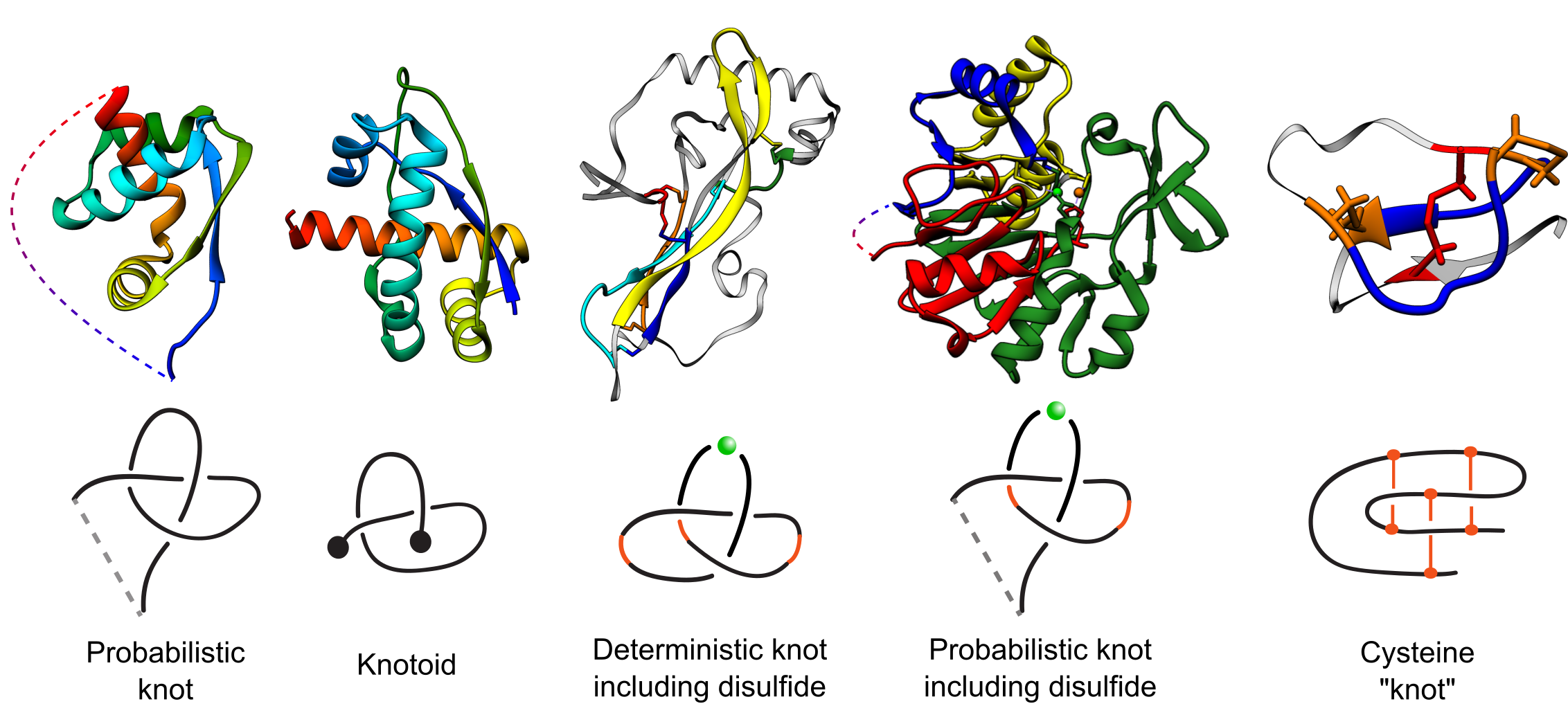
Fig. 1 Types of entanglements identified by the KnotProt 2.0. Probabilistic knots are identified based on a protein main chain (backbone). Deterministic knots are detected based on all possible combinations of disulfide or ion bonds with a protein backbone. In case of probabilistic disulfide and ion-based knots, the chain is closed based on the method established before for knotted proteins [1]. The knotoid analysis was achieved using the tools developed in [8].
More precisely, the novelties of KnotProt 2.0 (as compared to the first version of the KnotProt) include:
- it includes deterministic and probabilistic knots based on disulfide bonds, disulfide and ions bonds, and ions bridges
- it includes knotoids
- it includes cysteine knots
- the following filters are implemented: all or a non-redundant set of proteins, probabilistic knots (knots and knotoids), probabilistic knots including loops formed based on cysteines and ions, deterministic knots
- technical updates: a faster algorithm to detect non-trivial topology, more efficient algorithm to detect topological fingerprint
- all these novelties are also available via the server
The first examples of knots in proteins were found in 1994 [3], and many more have been identified in recent years [4, 5, 6]. Proteins may also form slipknots, i.e. contain knotted subchains even though their backbone chain as a whole is unknotted [1]; they were discovered in proteins in 2007 [2] and their systematic analysis was initiated in [7].
Moreover, knots can be also defined with disulfide bonds or ions taken into account [7]. Furthermore, non-trivial topology can be represented via so-called knotoids [8, 9]. Knotoids enable characterization of topology of open chains, such as represented by the majority of proteins, without requiring the endpoints to be connected in order to form a closed loop.
Usually it is impossible to determine, by a naked eye, if a given protein chain forms a knot (especially via cysteines or ions bonds), a slipknot, or a knotoid. Therefore, more involved mathematical tools, such as polynomial knot invariants, are used to detect such structures. Much effort has been invested into identifying all such entangled proteins among those deposited in PDB.
Recently considerable interest arose around this subject for a variety of reasons. First, it is believed that the presence of entangled structures in proteins is not accidental and therefore understanding their function is an important challenge. Second, recent work shows nearly perfect conservation of knotting fingerprints in some families whose members differ by hundreds of millions years of evolution (arising from distant organisms) and possess a low sequence identity [1]. Moreover, based on knotting fingerprints, it was shown that the locations of active sites in proteins are correlated with points characterizing their topology (e.g. positions of the knot core) [1]. These findings imply that a detailed representation of protein topology can be crucial for understanding their biological role. The KnotProt 2.0 database will make such data easily available and should help researchers to understand biological role of entangled proteins.
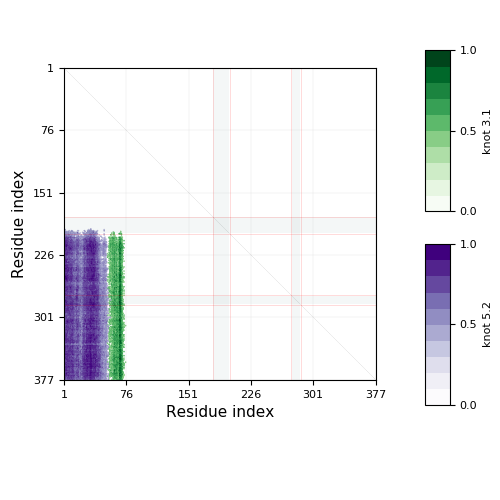
The database KnotProt 2.0 contains detailed information about the entanglement in proteins and presents it in the form of a "knotting fingerprint"
and the projection globes/maps
that present the data for the global topology (in the case of knotoids). The knotting fingerprint encodes information about the knot type of each subchain of a protein backbone and represents it in the form of a matrix diagram, see Fig. 1 and the detailed description in "Materials and methods”.
The KnotProt 2.0 database also presents extensive statistics about proteins with knots, knots defined with disulfide bonds or ions taken into account, slipknots, and knotoids based on their biological function, molecular tags, families association, type of fold, as well as geometric data: knotting patterns, knot and slipknot lengths and depths, etc. Interestingly, the KnotProt 2.0 analysis reveals that proteins with knots, slipknots, and knotoids can be classified into a few distinct topological motifs, represented by particular patterns within the matrix diagrams. This data can be used, for example, to find entangled proteins with a given homological sequence, a similar structure, or performing a particular biological function. Topological characterization of proteins rich in disulfide or ion bonds should help in explanation of their thermodynamical or mechanical stability, as well as reasons of misfolding. Moreover, proteins with cystein knots are the target of commercial and medical use (including anti-HIV, anti-bacterial and insecticidal activity). Representation of entanglement in proteins via knotoids shows not only the hidden complexity of proteins, but also gives a new interesting object to study for mathematician. The examples of possible application of topological characterization of proteins are given here . As an additional feature, a user can analyze structures and generate knotting fingerprints of uploaded proteins. It is also possible to upload and analyze a whole set of structures (e.g. analyze the evolution of a knot along a folding or unfolding trajectory). Details about the server option are described in Analyze your data
The KnotProt 2.0 database is automatically updated every Wednesday, immediately after new structures are deposited in PDB.
There are three main options users can choose from to view or analyze data:
- browse database, which enables users to browse all structures currently deposited in the database,
- search database, which enables users to classify proteins based on topology or different biological and sequential properties,
- process my structure, which allows to upload new polymer-like structures and analyze their topology, and to analyze time evolution of entangled structures.
After choosing or uploading some particular protein (or any other polymer chain), its knotting fingerprint is presented in the main window of the database. There are four options to choose from, displayed in blue in top of the window. The first option is Knotting data, which shows the topological structure, as described in detail in the “Knotting data” section. Other options ("Chain information summary", “Similar chains” (by sequence), and “Similar chains” (by structure)”) display useful information about proteins based in other biological databases (PUBMED, DOI, RCSB, PFAM); the key feature of this subpage is an automated classification of proteins based on the same knotting fingerprint, sequence similarity, structure and family origin.
[1] Sulkowska JI, Rawdon EJ, Millett KC, Onuchic JN and Stasiak A (2012)
Conservation of complex knotting and slipknotting patterns in proteins, PNAS 109, E1715–E1723.
[2] King NP, Yeates EO, Yeates TO (2007).
Identification of rare slipknots in proteins and their implications for stability and folding, J. Mol. Biol. 373, 153-66.
[3] Mansfield ML (1994)
Are there knots in proteins? Nat Struct Biol 1, 213–214.
[4] Taylor WR (2000)
A deeply knotted protein structure and how it might fold, Nature 406, 916–919.
[5] Virnau P, Mirny LA, Kardar M (2006)
Intricate knots in proteins: Function and evolution, PLoS Comput Biol 2, e122.
[6] Bölinger D, Sulkowska JI, Hsu H-P et al (2010)
A Stevedore's protein knot, PLoS Comput Biol 6, e1000731.
[7] Sulkowska JI, Sulkowski P, Onuchic JN (2009)
Jamming proteins with slipknots and their free energy landscape, Phys Rev Lett, 103, 268103.
[8] Goundaroulis D, Gügümcü N, Lambropoulou S, Dorier J, Stasiak A, Kauffman LH (2017)
Topological Models for Open-Knotted Protein Chains Using the Concepts of Knotoids and Bonded Knotoids, Polymers, 9, 444.
[9] Goundaroulis D, Dorier J, Benedetti F, Stasiak A (2017)
Studies of global and local entanglements of individual protein chains using the concept of knotoids, Scientific Reports, 7, 6309.
Protein chains in the database
The database contains almost every protein chain deposited in the pdb - redundant chains within particular pdb entry of homomultimeric complex are represented by one chain only. New pdb entries are checked each week.
We included non-X-ray entries and entries with Cα-only entries. Chains were subsequently evaluated to take into account insertions in these sequences of all non-typical aminoacids: MSE, FGL, LLP, SAC, SER, PCA, MEN, CSB, HTR, PTR, TYR, SCE, M3L, OCS, KCX, SEB, MLY, CSW, TPO, SEP, AYA, TRN. This analysis is performed so as not to introduce additional breaks along protein chain. In case of NMR structures, we took the first model with a given chain name. Out of those chains we identified around 1200 chains that possess either a knot or a slipknot. These currently comprise the KnotProt database.
The knotting fingerprint takes into account missing atoms, and in case they overlap with the knot core, they are represented by grey strips as in the figure below. In this case the missing part of the chain is replaced by a line segment. This may affect the type of knot detected, so one should be careful in interpreting results in such cases. Missing atoms in the chain are denoted in sequence representation in the "Knotting data" screen as ‘’-”.
KnotProt | Interdisciplinary Laboratory of Biological Systems
Modelling